

- Department of Neurology, Fattouma Bourguiba Hospital, Monastir, Tunisia
Correspondence Address:
Mariem Mhiri, Department of Neurology, Fattouma Bourguiba Hospital, Monastir, Tunisia.
DOI:10.25259/SNI_316_2024
Copyright: © 2024 Surgical Neurology International This is an open-access article distributed under the terms of the Creative Commons Attribution-Non Commercial-Share Alike 4.0 License, which allows others to remix, transform, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.How to cite this article: Mariem Mhiri, Amal Abbes, Rihab Ben Dhia, Narjes Gouta, Mahbouba Frih Ayed. Primary and metastatic cerebral Ewing’s sarcoma: A case report about a rare entity and literature review. 11-Oct-2024;15:367
How to cite this URL: Mariem Mhiri, Amal Abbes, Rihab Ben Dhia, Narjes Gouta, Mahbouba Frih Ayed. Primary and metastatic cerebral Ewing’s sarcoma: A case report about a rare entity and literature review. 11-Oct-2024;15:367. Available from: https://surgicalneurologyint.com/?post_type=surgicalint_articles&p=13148
Date of Submission
23-Apr-2024
Date of Acceptance
28-Jul-2024
Date of Web Publication
11-Oct-2024
Abstract
Background: Ewing’s sarcoma (ES) is a rare malignant tumor primarily affecting young individuals, with cranial localization being particularly uncommon. While intracranial metastatic ES is infrequent, only four cases of intracranial metastatic ES are reported in the literature; it presents unique diagnostic and therapeutic challenges.
Case Description: We present a distinctive case of ES to delineate its clinical, radiological, and histopathological characteristics. Our patient, a 33-year-old, manifested symptoms of intracranial hypertension and gait disturbance. Neurological examination revealed a static and kinetic cerebellar syndrome. Imaging studies and stereotactic biopsy confirmed the diagnosis of primary and metastatic cerebral ES. The treatment regimen encompassed chemotherapy and radiation therapy.
Conclusion: Our case underscores the importance of considering ES in the differential diagnosis of dural-based lesions exhibiting cystic components and heterogeneous contrast enhancement, particularly in young individuals. Early recognition and intervention hold promise for optimizing patient outcomes.
Keywords: Chemotherapy, Ewing’s sarcoma, Histopathology, Metastatic
INTRODUCTION
Ewing’s sarcoma (ES) is a malignant small round blue cell tumor primarily affecting bone and soft tissue, constituting approximately 4% of childhood and adolescent malignancies. While it predominantly manifests in bone, extraosseous ES involving the central nervous system (CNS) is rare. Only four cases of primary and metastatic cerebral localization have been documented, representing a mere 1% of all ES localizations [

CASE REPORT
A 33-year-old male presented with a gait disorder and acute headache accompanied by nausea, vomiting, and diplopia. He had no previous medical history. He had experienced persistent hiccups 2 weeks before admission. The endoscopy of the gastrointestinal tract was normal.
He consulted the Department of Neurology due to the persistence of headaches.
Neurological examination revealed signs consistent with a static and kinetic cerebellar syndrome and right abducens cranial nerve paresis. Ophthalmic examination demonstrated bilateral papilledema, while routine blood chemical analysis and complete blood count yielded normal results.
Magnetic resonance imaging (MRI) revealed multiple posterior and anterior cranial masses with heterogeneous enhancement involving the cerebellum and frontal lobe, featuring solid-cystic components. Bilateral transverse venous cerebral thrombosis was noted. Leptomeningeal enhancement was also noted [
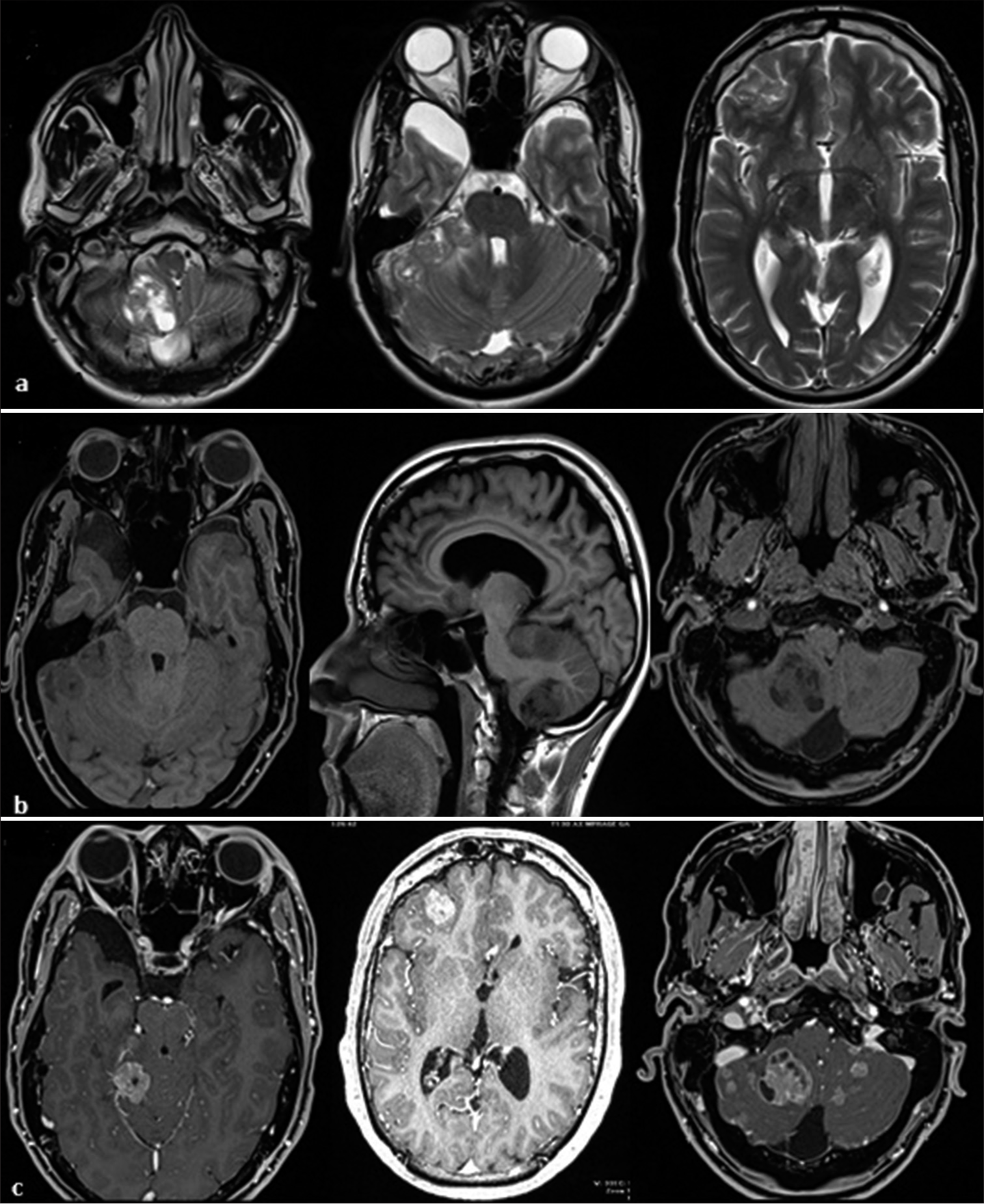
Figure 1:
(a) T2-weighted images: mixed hyper-intensity/hypo-intensity signals. (b) T1-weighted images: Extra-axial and intra-axial masses with no spontaneous hyperintensity and large area hypointensity. (c) T1 W images after gadolinium injection: solid cystic component mass with significant enhancement.
A stereotactic brain biopsy of the frontal lesion confirmed the diagnosis of ES. Subsequent spinal MRI revealed a metastatic intramedullary lesion on C4 and multiple intradural lesions on the thoracic spinal cord [

Histological examination demonstrated a highly cellular tumor comprising uniform small round cells with scanty cytoplasm, hyperchromatic nuclei, and frequent mitoses [

Immunolabeling was negative for CD45, glial fibrillary acidic protein (GFAP), chromogranin-A, and cytokeratins. The tumor cells were strongly and diffusely positive for CD99 with predominant membranous staining.
Considering the morphological, immunohistochemical, and molecular results, the final diagnosis was ES/primitive neuroectodermal tumor (PNET).
Due to the metastatic and multifocal nature of the disease, surgical intervention was not feasible. Instead, the patient received chemotherapy and focal irradiation, including six cycles of vincristine-etoposide-carboplatin alternating with vincristine-etoposide-cyclophosphamide, followed by curative radiotherapy with good clinical tolerance (60Gy over 30 fractions). Furthermore, he received three cycles of maintenance chemotherapy with cyclophosphamide and vinorelbine. Unfortunately, the patient died within 16 months of diagnosis.
DISCUSSION
ES is a rare family of malignancies in the CNS. Primary intracranial ES is extremely rare. It is the prerogative of children and young adults under 20 years old, with a peak incidence between 5 and 13 years old.[
Our case shows histological features similar to the few cases of CNS-ES previously described in the literature, but it displays different radiological aspects. In our case, masses are multiple and located infratentorial, extra, and intra-axial.
A review of the literature shows that the PNET is defined as an enhancing solid mass with hemorrhagic and cystic components.[
ES can be misdiagnosed as meningioma because it frequently arises from meninges. It manifests two different patterns of meningeal involvement characterized by diffuse involvement of leptomeninges or localized dural-based mass.[
ES mostly occurs as a single lesion. It is mostly intraparenchymal, located supratentorial or, less frequently, in the spinal cord.[
Immunohistochemistry for CD99 is characteristic of ES/PNET, but it is not specific because immunopositivity can also be expressed in other primary small cell tumors of the CNS. The pattern of staining is usually cytoplasmic in these tumors.[
However, central PNET is frequently negative for CD99 staining.[
It consists of the fusion of the human FLI gene on chromosome 11q24 with a gene EWS on chromosome 22q12, causing oncogenic conversion of the ES gene. It is found in over 90% of peripheral PNET-ES. The distinction of intracranial ES/PNET from central PNET is important for treatment and prognosis.
The therapeutic recommendation for ES indicates surgery when it is possible or combining systemic chemotherapy with focal irradiation.[
Drug combinations of chemotherapy include vincristineifosfamide-doxorubicin-etoposide therapy.[
The prognostic factors influencing intracranial extraosseous ES have not been identified yet. In general, a worse prognosis is associated with the presence of metastatic lesions at the time of diagnosis, infratentorial localization, involvement of bone, atypical histology, age at presentation >14 years old, and a tumor volume >200 mL.[
CONCLUSION
Primary and metastatic intracranial ES at diagnosis is exceedingly rare. Our case revealed the most atypical radiological features of all reported cases.
Physicians should consider the possibility of this rare tumor, especially in young people with dural-based lesions with cystic components and heterogeneous contrast enhancement. Histological and genetic features of the tumor are important for guiding treatment decisions and predicting prognosis.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Disclaimer
The views and opinions expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Journal or its management. The information contained in this article should not be considered to be medical advice; patients should consult their own physicians for advice as to their specific medical needs.
References
1. Batur A. Conventional and advanced MR imaging findings of primary Ewing sarcoma of the tentorium: A case report with literature review. Br J Neurosurg. 2021. 37: 1322-25
2. Cherif El Asri A, Benzagmout M, Chakour K, Chaoui MF, Laaguili J, Chahdi H. Primary intracranial pPNET/Ewing sarcoma: Diagnosis, management, and prognostic factors dilemma-a systematic review of the literature. World Neurosurg. 2018. 115: 346-56
3. Dedeurwaerdere F, Giannini C, Sciot R, Rubin BP, Perilongo G, Borghi L. Primary peripheral PNET/Ewing’s sarcoma of the dura: A clinicopathologic entity distinct from central PNET. Mod Pathol. 2002. 15: 673-8
4. Haveman LM, Ranft A, Van den Berg H, Klco-Brosius S, Ladenstein R, Paulussen M. Primary and metastatic intracranial Ewing sarcoma at diagnosis: Retrospective international study and systematic review. Cancers (Basel). 2020. 12: 1675
5. Huguenard AL, Li YD, Sharifai N, Perkins SM, Dahiya S, Chicoine MR. Multifocal primary central nervous system Ewing sarcoma presenting with intracranial hemorrhage and leptomeningeal dissemination: Illustrative case. J Neurosurg Case Lessons. 2021. 1: CASE2042
6. Ibrahim GM, Fallah A, Shahideh M, Tabori U, Rutka JT. Primary Ewing’s sarcoma affecting the central nervous system: A review and proposed prognostic considerations. J Clin Neurosci. 2012. 19: 203-9
7. Kazmi SA, Perry A, Pressey JG, Wellons JC, Hammers Y, Palmer CA. Primary Ewing sarcoma of the brain: A case report and literature review. Diagn Mol Pathol. 2007. 16: 108-11
8. Ke C, Duan Q, Yang H, Zhu F, Yan M, Xu SP. Meningeal Ewing sarcoma/peripheral PNET: Clinicopathological, immunohistochemical, and FISH study of four cases. Neuropathology. 2017. 37: 35-44
9. Roosen N, Lins E. Primary Ewing’s sarcoma of the calvarial skull. Neurochirurgia (Stuttg). 1991. 34: 184-7
10. Salunke PS, Gupta K, Malik V, Kumar N, Le H, Cai C. Primary Ewing’s sarcoma of cranial bones: Analysis of ten patients. Acta Neurochir (Wien). 2011. 153: 1477-85
11. Sohail AH, Sachal M, Maan MA, Soban M, Khan MS, Bari ME. Primary intracranial extraosseous Ewing’s sarcoma. Childs Nerv Syst. 2019. 35: 541-5
12. Tanboon J, Sitthinamsuwan B, Paruang T, Marrano P, Thorner PS. Primary intracranial Ewing sarcoma with an unusually aggressive course: A case report and review of the literature. Neuropathology. 2012. 32: 293-300
13. VandenHeuvel KA, Al-Rohil RN, Stevenson ME, Qian J, Gross NL, McNall-Knapp R. Primary intracranial Ewing’s sarcoma with unusual features. Int J Clin Exp Pathol. 2015. 8: 260-74
14. Weil RJ, Zhuang Z, Pack S, Kumar S, Helman L, Fuller BG. Intramedullary Ewing sarcoma of the spinal cord: Consequences of molecular diagnostics. Case report. J Neurosurg. 2001. 95: 270-5

